Genome sequencing has opened opportunities for detecting multiple variant types across the genome with a single technology. Previously, BCL has tackled evaluating copy number variant (CNV) detection from genome data using the DRAGEN™ 4.2.4 CNV pipeline. While CNV detection significantly expands the diagnostic power of genome data, there are still many known regions of the genome where sequence variant detection is difficult because of biological challenges. For example, many genes where variants are associated with genetic conditions and are reported in a clinical setting contain repetitive regions (e.g. NEB) or have high homology with a pseudogene (e.g. PMS2). These genes currently require specialized orthogonal assay design to detect variants with confidence.
With these challenges in mind, BCL has initiated efforts to evaluate new bioinformatic approaches to calling variants in these difficult genes. Illumina has designed specific targeted callers that can be run to detect sequence variants in these tricky regions using the DRAGEN software. Some of these callers are currently available, such as HBA caller that aids detection in the highly homologous genes HBA1 and HBA2, which are associated with α-thalassemia and the SMN1 caller, which detects copy number changes in the highly homologous genes SMN1 and SMN2, which are associated with spinal muscular atrophy.
This blog details the clinical need for these callers and introduces our latest effort in evaluating the performance of additional DRAGEN targeted callers, which will be detailed in subsequent blog posts.
Genome-wide tests make domain-specific expertise more difficult
With the decreasing cost of sequencing, Mendelian disease genes are being discovered more rapidly than ever before [1]. Per a policy statement of the American College of Medical Genetics and Genomics [2], all genes that have a gene-disease relationship of Moderate or above per the semi-quantitative framework developed by the Clinical Genome Resource (ClinGen) [3], are reportable in a diagnostic setting. As of December 2023, there were approximately 4000 genes in the Gene Curation Coalition database (search.thegencc.org) [4] that have a strength of Moderate or above and could be reportable on a clinical test.
If a clinical laboratory is going to offer a diagnostic test to report variants in a particular gene, the lab must understand all of the technical limitations of detecting variants in this gene. Depending on particular limitations, laboratories may be expected to develop multiple technologies to offer a highly sensitive and specific test [5]. For some genes, this ask is more difficult than others because of biology. This blog will speak to two different biological scenarios that often require special ancillary assays for a complete clinical test. Developing these assays takes time, can be costly, and often requires detailed knowledge of the gene, disease, and underlying biology of the system. This was more feasible when labs focused on small test menus and truly developed expertise in certain gene/disease areas, but as clinical laboratories try to move to genome-wide testing technologies, this task becomes exceedingly difficult.
Homologous Regions: Where is my variant?
Next generation sequencing (NGS) involves fragmenting genomic DNA into small pieces, usually ~350 base pairs each, ligating, barcoding, sequencing, and aligning them to a reference genome, much like putting together a jigsaw puzzle. The overall success of this method, particularly because the fragments are so short (usually ~150bp reads are generated), relies on the principle that many sections of the genome have a unique sequence. However, much like a puzzle that has certain colors or images where the pieces are indistinguishable, there are certain sections of the genome that are notoriously difficult to align. Regions of high homology fall into this bucket because they are regions in the genome that have near identical sequences (Figure 1). Homologous regions may have formed in evolution of the mammalian genome due to gene duplication events. One gene retains all of the functional elements of the active gene, while the other is an inactive gene copy, usually referred to as a pseudogene. Pseudogenes may contain all genetic sequence, or may be “processed” which means that all of the introns have been spliced out of that copy before it was reincorporated into the genome. Pseudogenes can be particularly subject to variation because of the lack of evolutionary constraint on the nonfunctional gene elements [6].
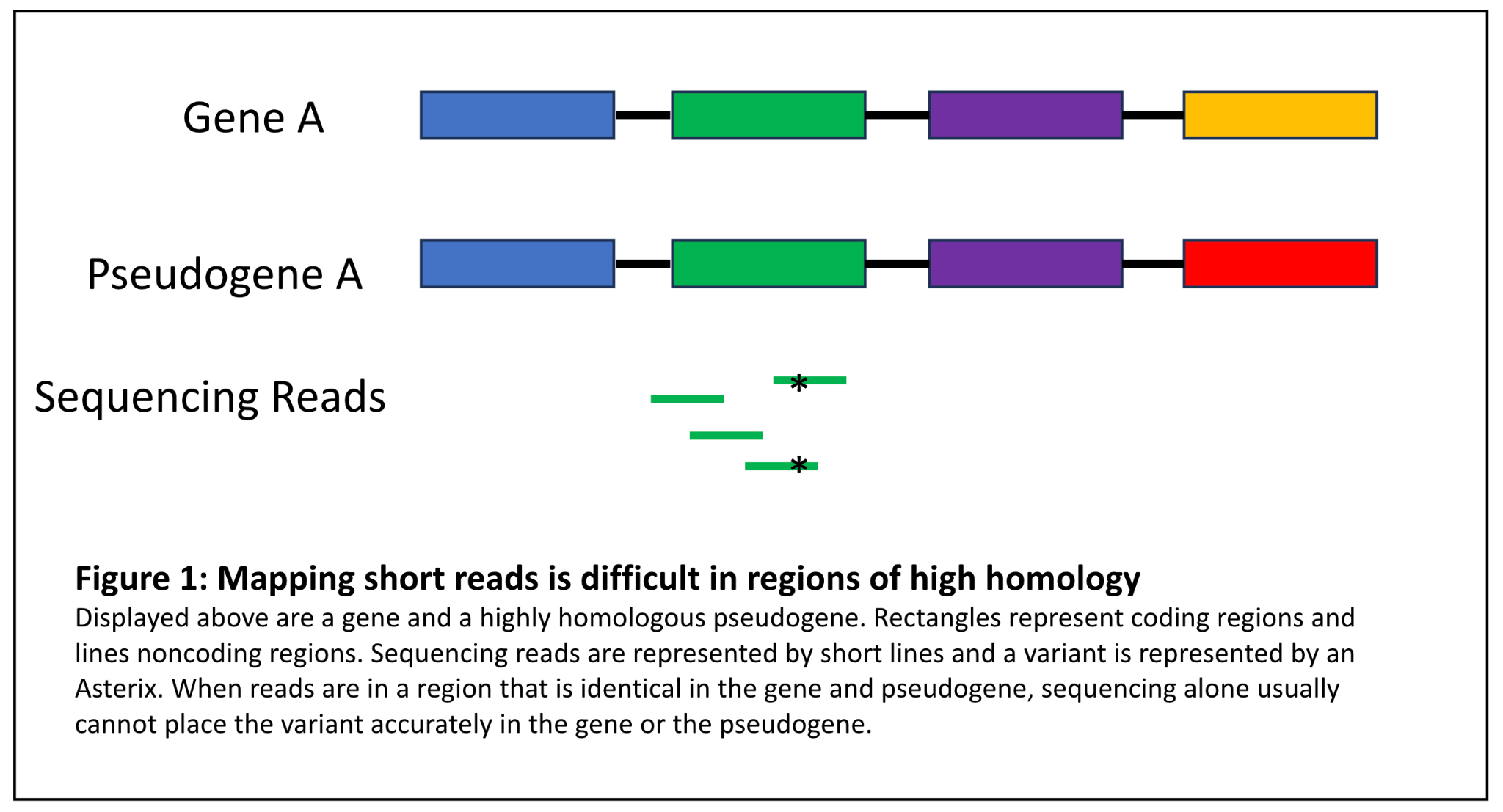
Figure 1: Mapping short reads is difficult in regions of high homology
Distinguishing between genes and pseudogenes can be critical because variants in some genes with a known pseudogene pair are associated with severe, highly actionable genetic conditions. This is illustrated by the fictitious case example below:
John Smith is a 45 year old patient with a history of colorectal cancer. Histology of a biopsy of this cancer reveals microsatellite instability (MSI). His clinical genetics team orders a comprehensive cancer panel test. The clinical lab detects a nonsense variant in exon 11 of PMS2. PMS2 is definitively associated with autosomal dominant Lynch syndrome, a syndrome with a high risk of early-onset colorectal cancer, among other cancers. The lab may have found the cause for John’s cancer. However, PMS2 has a highly homologous pseudogene PMS2CL that overlaps with exons 9 and 11-15 of PMS2 more than 98% identical sequence [7]. Before the lab can report this variant, they must determine if it is located in PMS2 (likely explains his condition, can inform familial cascade testing) or PMS2CL (pseudogene variant without clinical impact, no identified cause for his condition). NGS cannot differentiate between these regions; thus the laboratory must design and validate an ancillary assay to detect bona fide PMS2 variants in the pseudogene region. This usually consists of Multiplex Ligation-dependent Probe Amplification (MLPA), long-range PCR, or other methods. Reflexing to this assay when a variant is detected can add time and cost to a clinical sequencing test.
Differentiating between variants in genes and pseudogenes is also critical in a screening situation. If you alter the fictitious clinical scenario above slightly and say that John Smith is a 45 year old seemingly healthy individual who opted to do genetic screening and a nonsense variant was identified in exon 11 of PMS2, it would also be critical for the lab to determine if this variant were in PMS2 or PMS2CL to help inform screening protocols and prophylactic measures for this individual. If the variant were in PMS2, this individual would be at risk for Lynch syndrome cancers, but this would not be the case if the variant were in PMS2CL.
Repetitive Regions: Is my variant real?
A second bucket of difficult to align sequence in short read sequencing are those regions that contain long runs of repetitive material. If we return to the puzzle analogy, these would be sections in a puzzle that have a repeating pattern and the first part repeat of the pattern is not distinguishable from the third repeat of the pattern. It may be difficult to accurately detect variants in this region because the sequence is so repetitive (Figure 2). While it may not be as critical to place the variant in a repetitive region, it is critical to determine if a variant is actually present at all or is merely a sequencing artifact. This is illustrated in the fictitious example below:
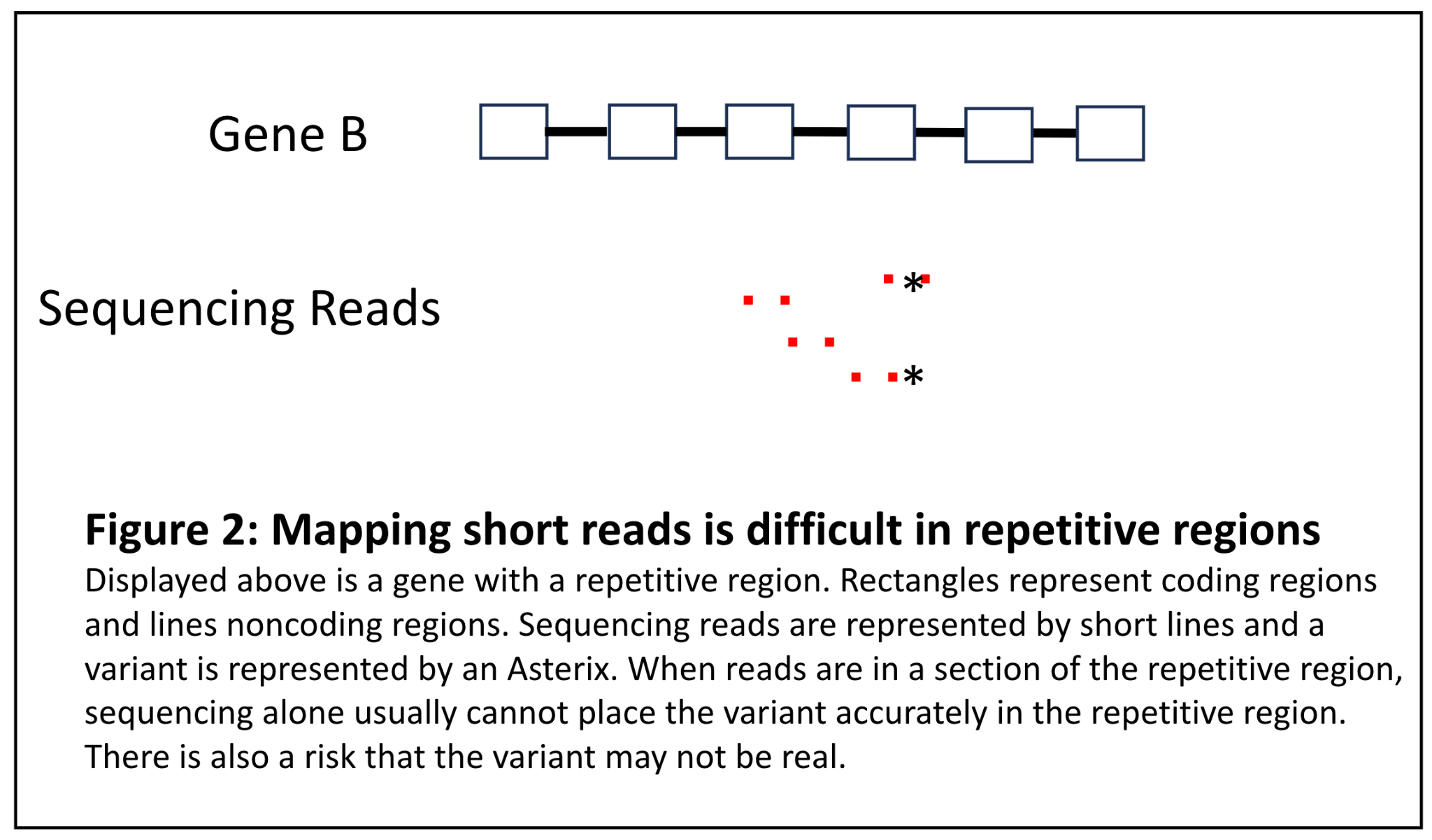
Figure 2: Mapping short reads is difficult in repetitive regions
Jane Doe is a patient with Nemaline myopathy and a muscle biopsy that has identified Nemaline rods. Her clinical team orders a comprehensive myopathy panel and a variant in NEB is identified. However, it is in the region of exons 82-105, which is a highly repetitive region that is a triplication of 8 exons [8]. The lab must determine if this variant is in fact real because it could be an explanation for Jane’s condition. However, because sequencing alone cannot determine this, an ancillary assay must be performed.
Future Directions
BCL, in collaboration with Illumina, has prioritized testing certain targeted callers that would help improve the sensitivity of our current genome sequencing product without the need to perform ancillary testing to detect variants in tricky regions. Current work is being done to source samples that have been tested with orthogonal methods to validate these callers. Future blog posts will be dedicated to the performance of new targeted callers for genes like PMS2, NEB, STRC, and HBA1 and 2, to name a few on the road map. Iterative development of bioinformatic algorithms to leverage the power of genome sequencing will help improve variant calling and, in turn, accurate clinical reporting from WGS, pushing it further toward that “one stop shop” genetic testing method of the future.
References:
1. Boycott, K.M., et al., International Cooperation to Enable the Diagnosis of All Rare Genetic Diseases. Am J Hum Genet, 2017. 100(5): p. 695-705.
2. Bean, L.J.H., et al., Diagnostic gene sequencing panels: from design to report-a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet Med, 2020. 22(3): p. 453-461.
3. Strande, N.T., et al., Evaluating the Clinical Validity of Gene-Disease Associations: An Evidence-Based Framework Developed by the Clinical Genome Resource. Am J Hum Genet, 2017. 100(6): p. 895-906.
4. DiStefano, M.T., et al., The Gene Curation Coalition: A global effort to harmonize gene-disease evidence resources. Genet Med, 2022. 24(8): p. 1732-1742.
5. Rehder, C., et al., Next-generation sequencing for constitutional variants in the clinical laboratory, 2021 revision: a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet Med, 2021. 23(8): p. 1399-1415.
6. https://www.ncbi.nlm.nih.gov/books/NBK535152/
7. Li, J., et al., A Comprehensive Strategy for Accurate Mutation Detection of the Highly Homologous PMS2. J Mol Diagn, 2015. 17(5): p. 545-53.
8. Yuen, M. and C.A.C. Ottenheijm, Nebulin: big protein with big responsibilities. J Muscle Res Cell Motil, 2020. 41(1): p. 103-124.
9. Blog Thumbnail Photo by Warren Umoh on Unsplash
Important note for this blog: Posts do not equal endorsements. Opinions expressed in this blog are those of the author, on behalf of the genomics group at Broad. We make every effort to ensure the accuracy of data/figures presented here but these are not peer-reviewed and errors may occur from time to time. Broad has a collaboration agreement with Illumina that in-part funds this work.