Copy Number Variations (CNVs) are pivotal in shaping human genetic diversity and influencing disease susceptibility. These segments, either deleted (DEL) or duplicated (DUP), can dramatically alter gene dosage, thereby modulating phenotypic outcomes1. Given their impact, the precision of CNV calling is not just a technical requirement but a critical factor for robust downstream analyses and accurate genetic interpretation. We are interested in evaluating the CNV calling capabilities of the latest available DRAGEN™ version with an eye toward future validation and inclusion in Broad’s existing clinical WGS pipeline (which currently uses DRAGEN™ v3.10.4).
A significant enhancement in the DRAGEN™ 4.2.4 CNV calling pipeline is that CNV calls now come with SV support. In previous versions, DRAGEN™ CNV callers primarily rely on depth signals for detecting CNV events. The underlying concept behind this approach is that short reads are randomly sampled on the genome, and copy number can be inferred from the read depth of aligned short reads2. However, while depth signal is reliable for detecting large CNV events, its efficacy diminishes significantly when dealing with shorter CNV events (<10kbp) due to a lower signal to noise ratio. Consequently, many smaller CNV events were historically left to be identified by the DRAGEN™ structural variant (SV) caller, which utilizes junction signals. In practice, this presented a challenge as researchers needed to perform an extra processing step to merge CNV events from both DRAGEN™ SV and CNV callers’ outputs, potentially introducing inaccuracies. In the latest iteration, DRAGEN™ 4.2.4 introduced an additional integration step that leverages junction signals from the SV caller and depth signal from CNV caller to generate the cnv_sv.vcf. This newly introduced VCF includes a SVCLAIM field which indicates how each CNV event was detected – whether through depth signal D, or junction signals J, or a combination of both DJ. Illumina has indicated that this improvement streamlines the workflow for researchers and boosts the accuracy of CNV calls, especially with shorter CNV events (<10kbp)3.
This blog introduces our latest effort in evaluating the performance of the DRAGEN™ v4.2.4 CNV caller with SV support. We have used the conventional method of comparing a query VCF file to benchmarking a VCF file which helped us in identifying true positives (TPs) when query variants matching events in benchmark datasets. However, we have found limitations of currently available benchmarking VCFs, often omitting relevant events, which hinder our ability to accurately estimate false positive rates – a pivotal metric in our clinical validation. Recognizing these limitations, we have developed a more robust and reliable alignment-based evaluation method to address shortcomings in the conventional approach while enhancing our understanding of CNV events.
Old School: Conventional CNV Caller Evaluation
Traditionally, we utilize benchmarking datasets to evaluate the performance of our variant calling pipelines. The National Institute of Standards and Technology (NIST) has provided a benchmark ‘truth’ dataset for the HG002 genome, generated using dipcall – a reference-based variant calling pipeline. Dipcall employs the HG002-T2T (Telomere-to-Telomere) v0.9 assembly as its ground truth. The T2T assembly represents one of the most accurate genome assemblies4,5 and offers a comprehensive view of CNV events. The assembly is then compared against the hg38 reference genome to generate a robust and precise variant benchmarking dataset for HG002. However, it’s important to note because dipcall relies on the hg38 genome as a template for variant calling, the output may be biased against some unresolved regions in hg38. Because long-read technology has superior capabilities in detecting structural variants, we also use PBSV calls from long-read HiFi data of the hg38 genome as another means for benchmarking accuracy on HG0026. It’s important to recognize this dataset, like the NIST benchmarking dataset, may have its own pitfalls. HiFi has its own limitation in detecting extra-long CNV (>20kbp) events. Those benchmarking datasets allow us to measure the accuracy of DRAGEN™ CNV calling pipeline. We have conducted a comparative analysis using Witty.er by comparing the results of DRAGEN™ v3.10.4 CNV calls, DRAGEN™ v3.10.4 bcftools concat CNV SV calls, and DRAGEN™ 4.2.4 CNV_SV calls against the HG002 dipcall and HiFi PBSV benchmarking datasets within the high-confidence region7.
As illustrated in Figure 1, DRAGEN™ 4.2.4 demonstrates a strong correlation with dipcall and HiFi benchmarking datasets, suggesting its proficiency in identifying CNV DEL events across various event lengths, especially those under 10 kbp threshold. On an event-level assessment, the performance of the 4.2.4 cnv_sv caller is closely aligned with 3.10.4 bcftools merged calls. However, it’s worth noting that performance outcomes exhibit variations depending on the choice of benchmarking datasets. For instance, when using dipcall as benchmarking reference, DRAGEN™’s precision in the 20-50kbp and >50 kbp is notably higher than when utilizing HiFi PBSV as the benchmarking dataset. One possible explanation for this is the limitation of HiFi in detecting extra-long CNV events. Our results highlighted DRAGEN™ CNV caller performance and the impact of selected benchmarking datasets on performance evaluation.
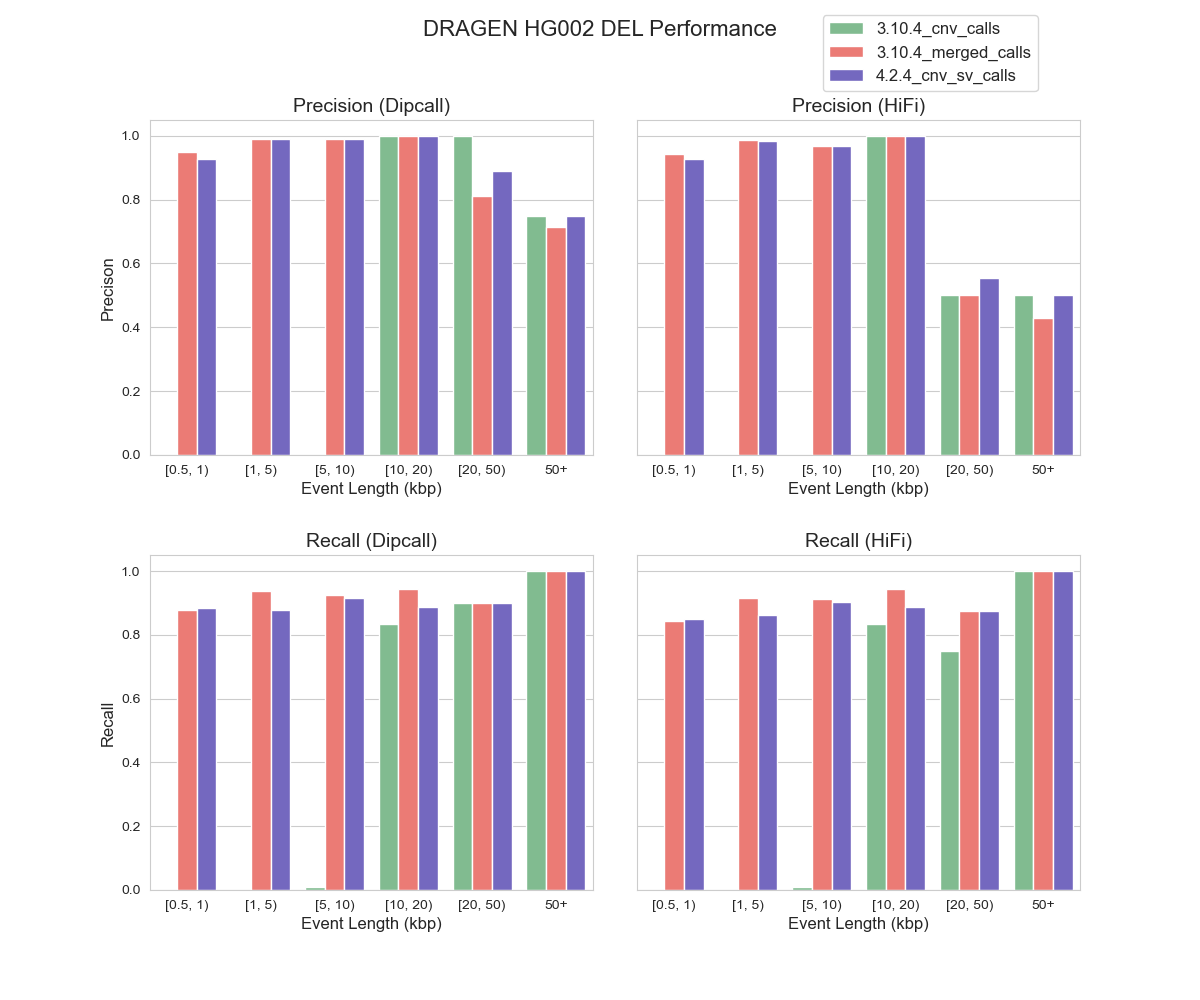
FIGURE 1. Precision and Recall of DEL Events by DRAGEN™ CNV in HG002 (hg38) Using Dipcall and HiFi PBSV as Benchmarking Datasets
For CNV detection, the task of identifying DUP events has long been acknowledged as more challenging as compared with identifying DEL events. This challenge can be attributed to lack of sensitivity of both depth signal and junction signal in distinguishing a change in copy number accurately8. Consequently, neither of the benchmarking datasets we use differentiate between insertions and duplications, which poses a challenge in assessing the performance of DRAGEN™ caller for DUP events. To address this limitation, we used an external tool SVWIDEN to annotate DUP events within the benchmarking datasets. Given the complexity of DUP event detection, it came as no surprise that our DUP events evaluation yielded less ideal results (Figure 2). Moreover, we also observed performance discrepancies across different benchmarking datasets pointing to the incompleteness of our benchmarking datasets for DUP events. This prompted us to explore an alternative alignment-based method to evaluate CNV events.
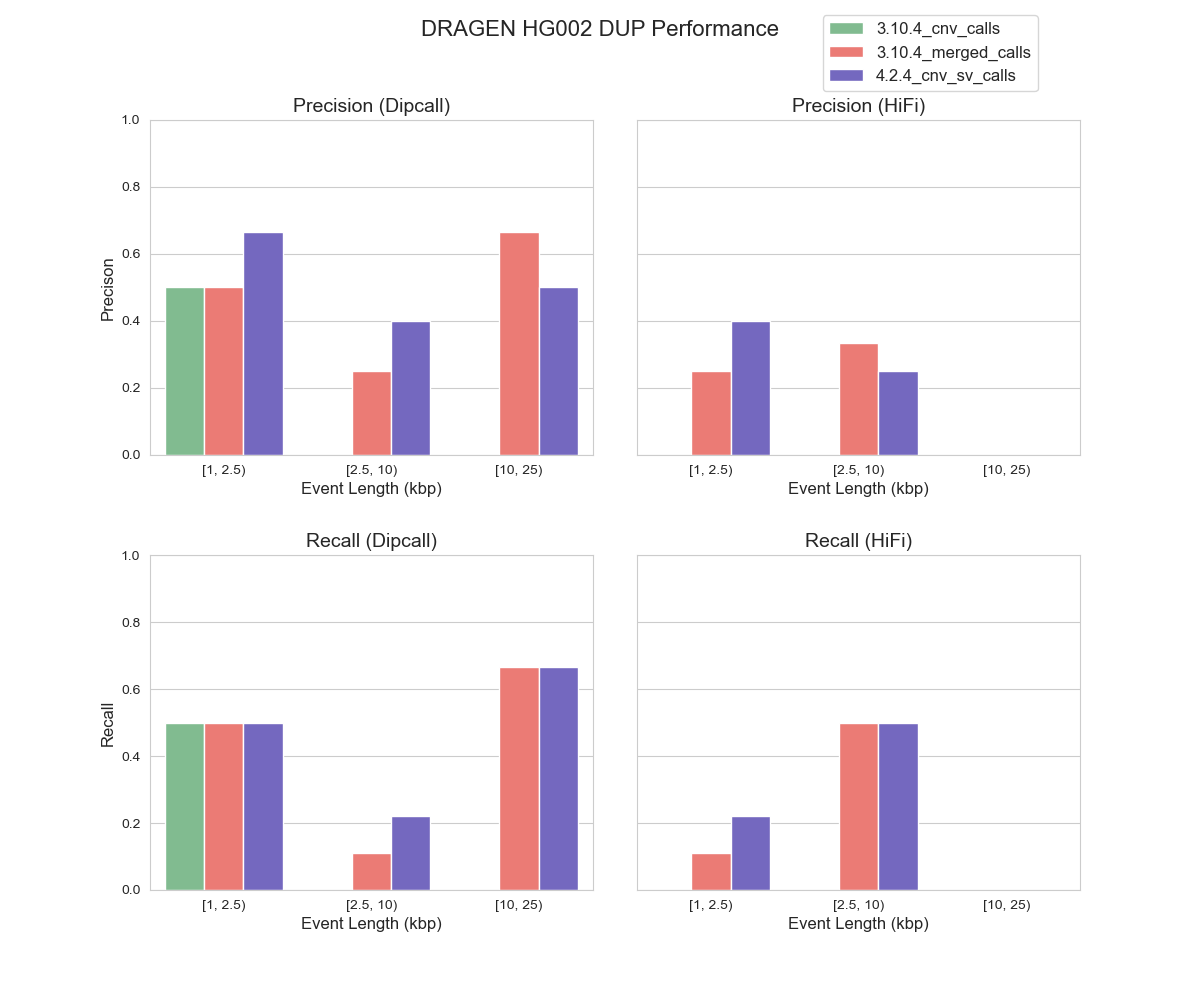
FIGURE 2. Precision and Recall of DUP Events by DRAGEN™ CNV in HG002 (hg38) Using Dipcall and HiFi PBSV as Benchmarking Datasets
A New Approach: Alignment-Based CNV Evaluation
In this method, we use an alignment search tool (LAST9) to identify CNV event sequences relative to both the HG002-T2T v0.9 assembly and the hg38 reference genome. We align the DNA sequence representing a DUP variant called in hg38 to the HG002-T2T reference. A TP DUP event is characterized by a higher copy number in HG002-T2T than in hg38, while a FP DUP event shows fewer copies in HG002-T2T than hg38. Since the hg38 assembly is haploid, and the HG002-T2T assembly is diploid, we assume a copy neutral event has one copy in hg38 and two copies (one for the maternal and paternal haplotypes) in HG002-T2T. A copy is defined as a LAST alignment where the alignment length is at least 95% of the query length and the percent identity of the alignment is at least 95%. This method allows us to identify not only whether a CNV event is correct, but in the case of duplications also allows us to identify the precise number, locations, and genotype of duplication events, even if they occur on separate chromosomes from the original call on hg38.
By using this method, we scrutinized certain DUP events that were absent in both HG002 benchmarking datasets we used. Our objective was to figure out whether these events were indeed false positives (FPs) or if their classification was a byproduct of the limitations inherent in the benchmarking datasets we relied upon. As shown in Figure 3B, a specific DRAGEN™-called DUP event with depth signal on chromosome 10 (chr10) attracted our attention. We identified seven copies on HG002-T2T’s chr10 in the maternal side and one copy on the paternal side. However, only one copy exists in the corresponding region in the hg38 genome. Although this DUP event was not present in either the dipcall or HiFi PBSV VCF, our alignment results strongly suggest that the genomic region chr10:39364454-39376272 in hg38 indeed represents a genuine DUP event in HG002. Furthermore, our alignment-based evaluation method unveiled a valuable insight into the mechanism behind this DRAGEN™-called DUP event, revealing it as a heterozygous tandem duplication event.

FIGURE 3. Detailed Analysis of a Unique DUP Event. A. An Integrated Genomics Viewer (IGV) screenshot displays a genomic region of interest. The highlighted red region represents a DUP event uniquely called by DRAGEN™, and absent in both HG002 dipcall VCF and HiFi PBSV VCF benchmarking datasets. Additionally, two green-highlighted DEL events are observed, with their benchmarking variants present in HG002 dipcall VCF but not in HiFi PBSV VCF. B. The alignment search results using LAST for the red-highlighted DUP event in 3A. Copies of this event are shown in hg38, HG002 maternal, and HG002 paternal chromosomes.
Obtaining a precise depiction of the genomic landscape of CNV events is a challenging quest. Our alignment-based evaluation method has proven itself as a powerful tool, shedding light not only on individual DUP events but also on the intricate relationships between multiple CNV events. Figure 4A depicts two DRAGEN™-called DUP events that were positioned approximately 70 kbp apart from each other. Neither DUP events were found in the benchmarking datasets that we used. What amplifies the intrigue is our discovery regarding DUP event a (chr1:16605769-16645359) and DUP event b (chr1:16715827-16727637), challenging the conventional spatial arrangement depicted in hg38 coordinates. According to hg38 coordinates, DUP event a occurred before DUP event b. However, our alignment results reveal a different scenario: on both the maternal and paternal chromosomes of HG002-T2T DUP event b precedes event a in sequence (Figure 4B). This intriguing twist is accompanied by a consistent presence of two copies on the maternal side and three copies on the paternal side, suggesting both DUP events are TPs and might be located on the same haplotype. Furthermore, our method reveals these copies to be dispersed, suggesting both events are homozygous dispersed DUP events. Our alignment-based CNV evaluation method not only unveils the intricate details of DUP events but also illuminates their spatial distribution, expanding our understanding of the complexity of CNV events.
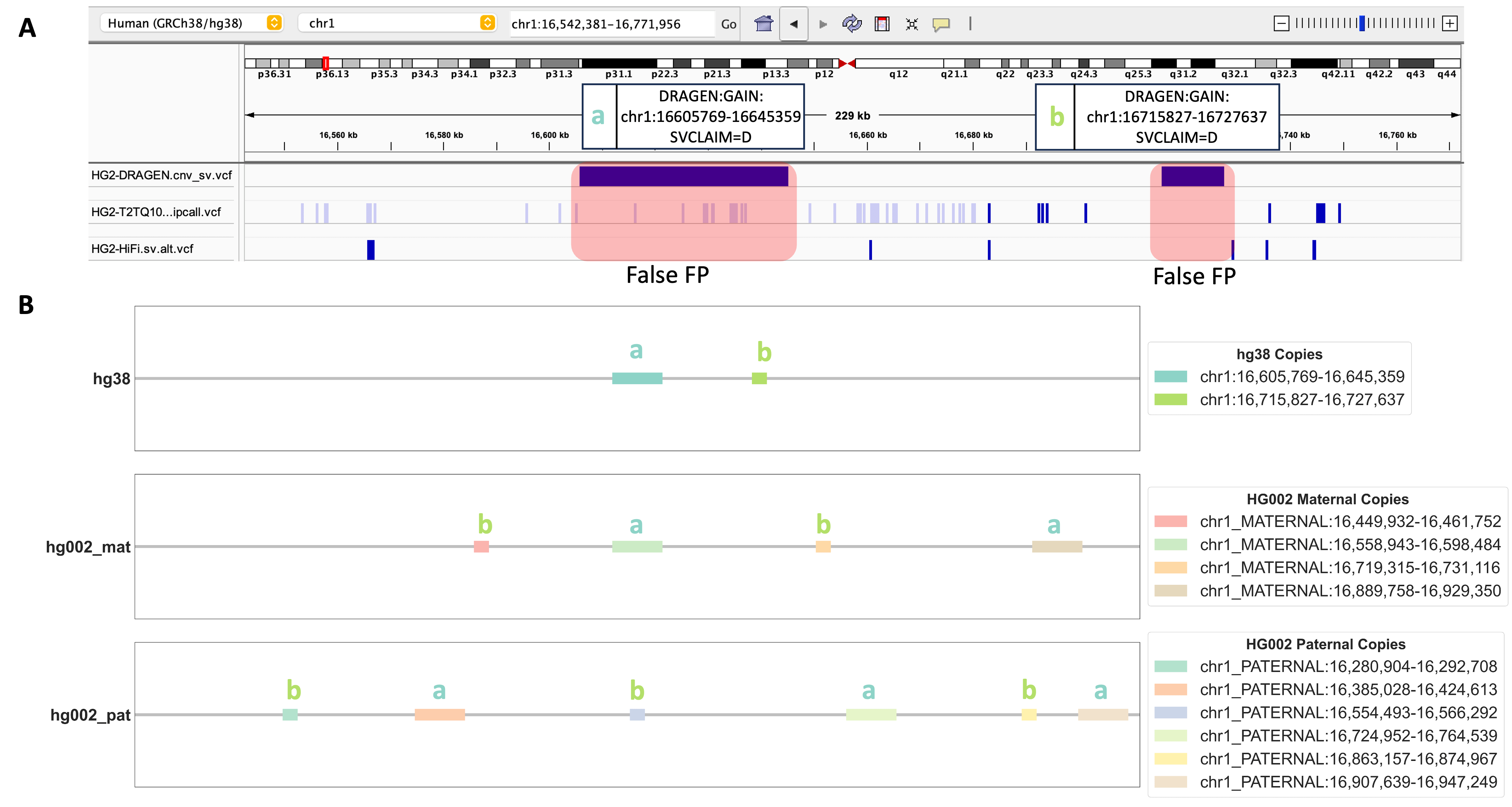
FIGURE 4. Investigation of Two Adjacent DUP Events Identified by DRAGEN™. A. An IGV screenshot displays the genomic region containing DRAGEN™-called DUP event a (chr1:16605769-16645359) and DUP event b (chr1:16715827-16727637). Three genomic tracks from top to bottom include DRAGEN™ 4.2.4 output cnv_sv.vcf, HG002-T2T v0.9 dipcall VCF, HG002 HiFi PBSV VCF. B. Alignment results for DUP event a and DUP event b. Copies of DUP event a and DUP event b are shown in hg38, HG002 maternal, and HG002 paternal chromosomes.
In addition, we confronted an interesting case involving a DUP event located at chr17:22130212-22156606, a genomic region that also lacked any support from the benchmarking datasets we relied upon (Figure 5A). Unfortunately, we were unable to find supporting evidence for this 26kbp DUP event using our alignment-based method, which only had depth-only signal (SVCLAIM=D). As shown in Figure 5B, we observed only one copy of this sequence within the HG002 maternal chromosome, conspicuously absent on the HG002-T2T’s paternal chromosome. This observation defied our hypothesis, as we anticipated a DUP event sequence should have two or more copies of this sequence on either maternal or paternal chromosome. The presence of just a single copy on the HG002-T2T maternal chromosome strongly points to a putative false positive DUP event. It’s worth noting that our alignment-based evaluation method imposes stringent requirements on the precision of genomic coordinates. Thus, while it remains plausible that this genomic region harbors a genuine DUP event, its event length might be smaller than what’s indicated by DRAGEN™ CNV calling algorithm.
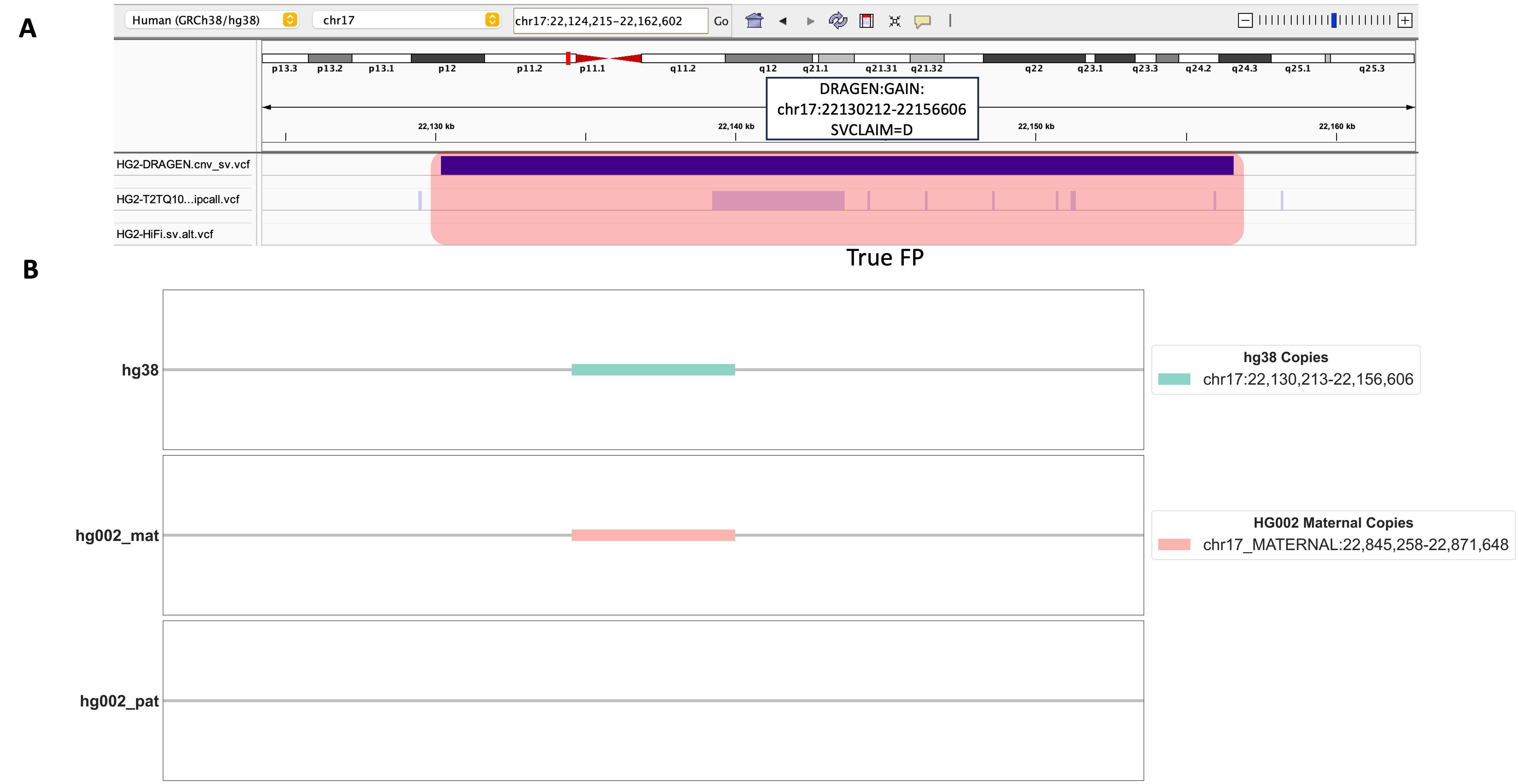
Figure 5. A Detailed Investigation of a DUP Events Identified by DRAGEN™ CNV. A. An IGV screenshot displays the genomic region harboring one DRAGEN™-called DUP event chr17:22130212-22156606. Three genomic tracks from top to bottom include DRAGEN™ 4.2.4 output cnv_sv.vcf, HG002-T2T v0.9 dipcall VCF, HG002 HiFi PBSV VCF. B. Alignment results for DRAGEN™-called DUP Event chr17:22130212-22156606. Copies of this DUP event are shown in hg38, HG002 maternal, and HG002 paternal chromosomes.
Overall, our alignment-based CNV evaluation method represents a pioneering approach in the intricate realm of CNV events. By harnessing the power of the HG002-T2T genome assembly, we’ve unlocked the ability to not only unveil CNV events in finer resolution but also delve deeper into spatial relationships and characteristics of adjacent CNV events.
Conclusion and Future Work
In this work, we evaluated the performance of the DRAGEN™ v4.2.4 CNV caller, utilizing dipcall events from HG002-T2T v0.9 assembly and HiFi PBSV data as our benchmarking datasets. It’s worth noting that some of the FPs initially identified using this method ended up revealing limitations within the benchmarking datasets. The limitations inherent in conventional evaluation approaches prompted us to embark on a journey towards developing a new alignment-based methodology by harnessing the power of the HG002-T2T genome assembly. Using this novel approach, we found some of the query FPs suggested by benchmarking datasets were actually TP events, highlighting the room for improvements of benchmarking datasets. Conversely, some FPs were indeed true FPs providing valuable insights that can inform methods or filtering strategies in future CNV callers.
While we have made significant strides in understanding CNV events within the HG002 sample, we acknowledge that a full validation across all DRAGENTM called CNV events leveraging our alignment-based approach is yet to be completed. As we anticipate a growing number of completed genomes emerging from the T2T effort, we hope to streamline and refine this alignment-based CNV evaluation method, providing a transformative means of evaluating CNV events across a broader spectrum of genomes.
References
- Ruderfer, et al. Patterns of genic intolerance of rare copy number variation in 59,898 human exomes. Nat. Genet, 2016
- Yoon S, et al. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res, 2009
- https://www.illumina.com.cn/content/illumina-marketing/spac/en_AU/destination/Webinar-Illumina-DRAGEN-4-2-Enhanced-machine-learning-new-targeted-callers-and-more.html
- Wang T, et al. The Human Pangenome Project: a global source to map genomic diversity. Nature, 2022
- Rautiainen M, et al. Verkko: telomere-to-telomere assembly of diploid chromosomes. Nature, 2022
- Zook J, et al. A robust benchmark for detection of germline large deletions and insertions. Nature Biotech, 2022
- Ebert, Peter, et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science, 2021
- Teo SM, et al. Statistical challenges associated with detecting copy number variations with next-generation sequencing. Bioinformatics, 2012
- Kielbasa, et al. Adaptive seeds tame genome sequence comparison. Genome Res, 2011