At the Methods Development Lab (MDL) of Broad Clinical Labs we are always looking to innovate on the latest genomics technologies. One promising new approach is from Fluent BioSciences for single-cell RNA sequencing. Their PIPseq platform can rapidly generate single-cell emulsions by vortexing a cell suspension with particles they call PIPs (Particle-templated Instant Partitions)1. This technology has the potential to generate very large single-cell libraries at a low cost per cell. Given these favorable characteristics, we were keen to investigate PIPseq’s compatibility with our recently developed high-throughput RNA isoform sequencing approach, Multiplexed Arrays Sequencing (MAS-seq). Together, these approaches could enable the large-scale measurement of isoform expression in 10⁴ to 10⁶ cells.
MAS-seq works by concatenating a defined number of cDNAs, typically 16, into long single molecules, termed MAS arrays2. The length of the MAS arrays are tailored to be in the optimal size range for the long-read PacBio sequencing platforms, approximately 10-18kb. After sequencing, the MAS arrays are deconcatenated informatically, boosting sequencing output by the number of constituent cDNAs per array. PacBio has commercialized MAS-seq and provides kits that boost output for bulk and single-cell RNA sequencing libraries by 16 fold.
The current MAS-seq protocols were developed using 10x Genomics Single-cell Gene expression libraries. In theory, MAS-seq is compatible with any method that generates full-length cDNA molecules and contains the appropriate PCR adapter handles. However, unforeseen obstacles can arise when integrating protocols. In this post, we demonstrate successful integration of MAS-seq and PIPseq in the context of peripheral blood, and show that the combination of both technologies provide high-quality single-cell isoform data.
Experiment
To test out this new method we looked at a classic single-cell sample: peripheral blood mononuclear cells (PBMCs). We used Fluent’s T20 3′ kit to encapsulate single cells into droplets according to the manufacturer’s protocol. After single-cell library construction, we had a pool of full-length cDNA molecules with barcodes and UMIs attached. Here, we separated the full-length cDNA library into two parts: one continued with the standard protocol and went on an Illumina sequencer to get short read gene expression data. The other pool was used as input for MAS-seq.
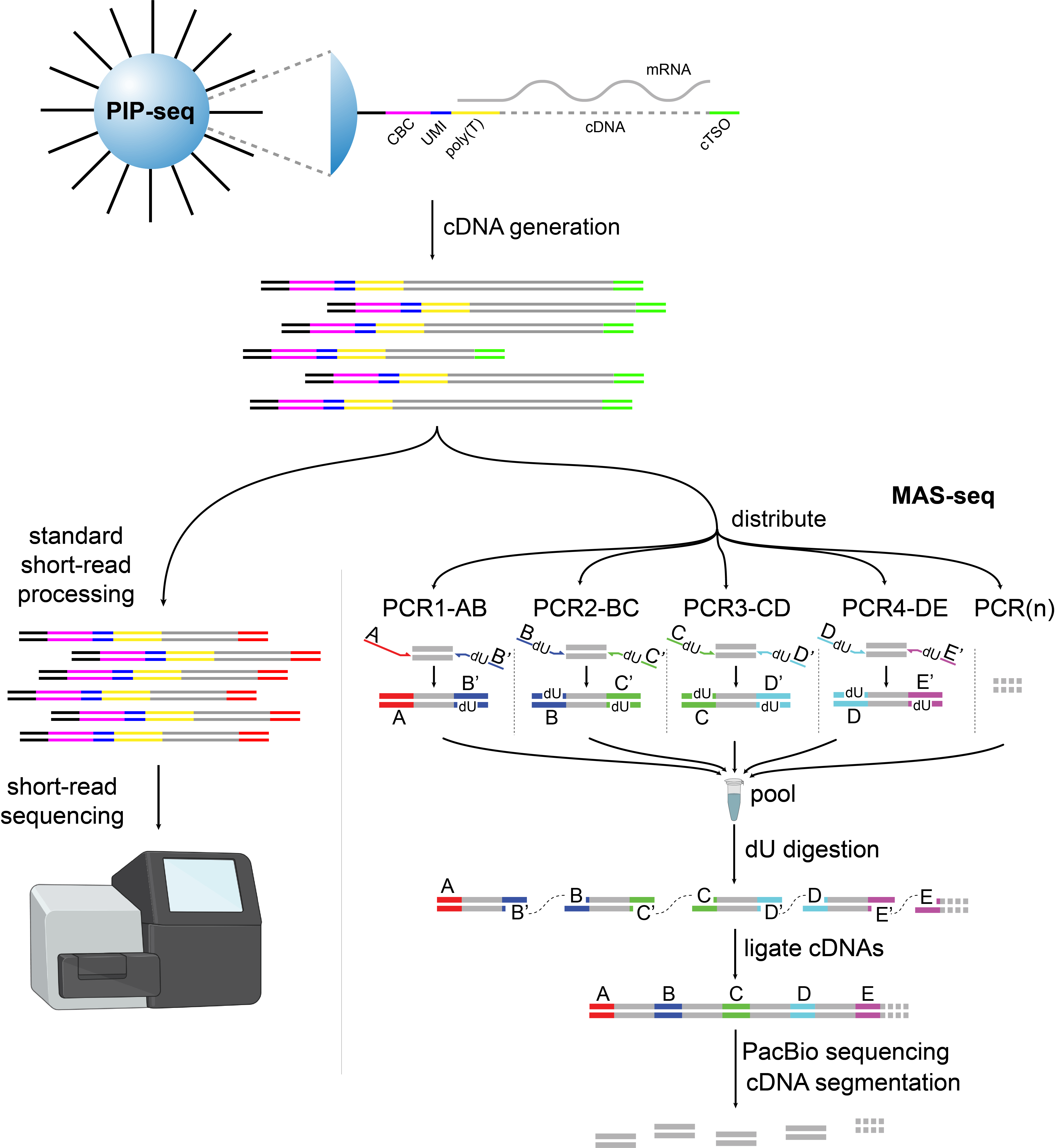
Figure 1: Overview of the experimental workflow. PBMCs were encapsulated into droplets with the PIPseq T20 3′ kit. After cDNA synthesis the pool was split in two and input to both short- and long-read protocols. Illustration created with BioRender.com.
Short-read PIPseq
First we examined the short-read data that we generated from the Fluent platform. We sequenced the library on a NovaSeq X 10B, generating 1.1 billion 246-bp cDNA reads. We used Fluent’s computational pipeline, called PIPseeker, to assign reads to barcodes, count UMIs, and map to the genome. The result is a familiar MTX-formatted file of UMI counts, along with the list of barcodes and features. These files are compatible with the standard tools and processes for analyzing single-cell sequencing data.
To identify cells, we selected cell barcodes containing a minimum of 1,000 UMIs and between 2-7% mitochondrial reads (Fig 2). After applying these filtering steps, 7,029 cell barcodes remained.
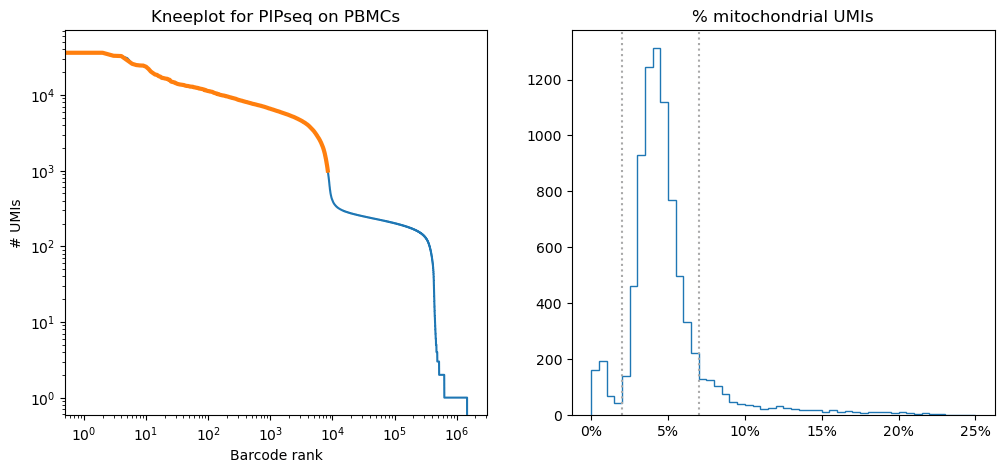
Figure 2: Left: knee plot showing the distribution of UMIs per barcode sorted in descending order. Selected barcodes are highlighted in orange. Right: histogram of the percent mitochondrial UMIs per cell, with dotted lines showing the cutoffs used to select barcodes.
After performing gene selection and Leiden clustering, we identified eight canonical cell types that should be familiar to people who work with peripheral blood. There is certainly more complexity in this dataset and further clustering would resolve some of the structure visible, but for our purposes it was sufficient to identify the major cell types in the data.
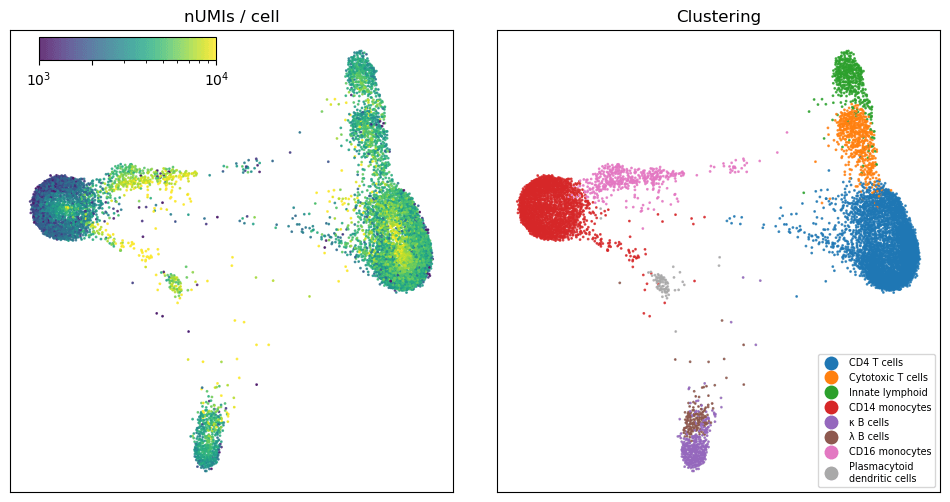
Figure 3: Graph embedding of short read data. The plot on the left shows the number of UMIs per cell, on a log scale. On the right the cells are colored by cell type.
Consistent with the described behavior of PIP-seq’s V4 chemistry, we saw lower counts in the CD14 monocytes than in other cell types3. This is a known challenge when processing cells that express high levels of RNAses, which is the case in the CD14 monocyte population.
Full-length cDNA using PIPseq
We next sought to determine if cDNA generated by PIPseq is compatible with MAS-seq for single-cell isoform-specific long reads. We split a portion of the pool of PIPseq full-length cDNA to be used as input into the MAS-seq library prep kit from PacBio (cat# 102-659-600) to generate 16-mer cDNA arrays.
After array creation, we sequenced the MAS-seq library on a PacBio Revio, yielding 3,062,399 HiFi reads. After segmentation with PacBio’s tool skera, we ended up with 46,116,893 s-reads, demonstrating a >15-fold boost in output. A total of 2,744,221 reads (89.6%) of the HiFi reads were full-length arrays, with an average length of 11,889 bp (Fig 4). The segmented reads had an average length of 725 bp.
We extracted the barcodes and UMIs from these reads, and mapped the long-read cDNA to the same genome that we used for the short-read data. Following barcode extraction and mapping, we quantified isoforms with IsoQuant and associated those calls with the Fluent barcodes to get a cell-by-isoform matrix4.
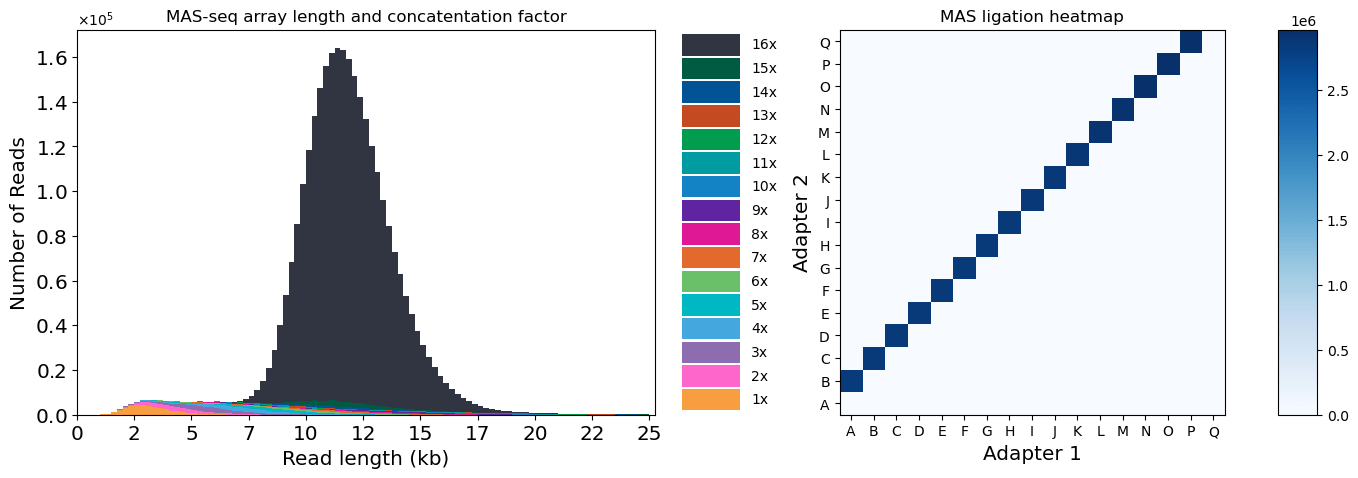
Figure 4: Quality control for MAS-seq data. Left: concatenation factor and read length distribution for HiFi reads. Right: heatmap of ligation adapter pairs.
When we looked at our long-read data we found nearly all of the cell barcodes that we selected in the short-read data: 7,023 out of 7,029, or 99.9%. Consistent with the short-read data, long-read counts for CD14 monocytes were considerably lower than for other cell types. This effect was potentially exacerbated by our long-read processing protocols that preferentially filter out shorter and likely degraded cDNAs.
| Reads after segmentation | 46,116,893 |
| Bead barcodes in whitelist | 498,380 |
| Reads with barcodes in whitelist | 30,766,203 |
| Total UMIs, all barcodes | 18,160,470 |
| Total UMIs, selected cell barcodes | 12,834,983 |
| Median UMIs/cell | 1,253 |
| Median isoforms/cell | 462 |
Table 1: Key metrics describing the single-cell MAS-seq results
Isoform-level quantification in PIPseq
While RNA degradation is apparent in the CD14 monocytes, the other cell types were sequenced at high depth. We found many isoforms to be specifically or preferentially expressed in specific cell types, recapitulating the findings of Inamo et al., 2022 and other groups5. Below we show two examples.
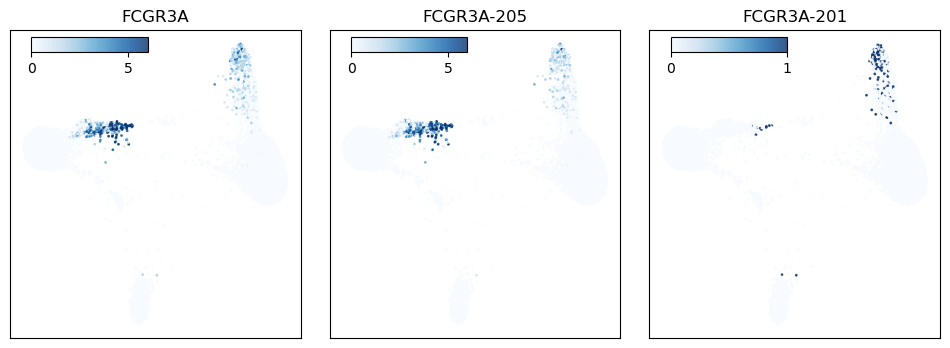
Figure 5: FCGR3A expression plotted across the cell population. The left panel shows FCGR3A gene expression, while the middle and right panels show FCGR3A isoform expression.
Example 1: FCGR3A (CD16)
FCGR3A (CD16) is an Fc receptor gene and a canonical marker of non-classical monocytes. When the protein is expressed on the cell surface, it serves as a receptor for IgG antibodies and triggers antibody-dependent cell-mediated cytotoxicity. At the gene level FCGR3A is highly expressed in the associated monocyte population, with lower expression in cytotoxic T cells and innate lymphoid cells (Fig 5). When we look at the data at the isoform level, we see that the latter two cell types express an alternate isoform that is absent from the CD16 monocyte cluster. This isoform has an additional exon in the 5′ UTR, suggesting that its inclusion may serve a regulatory role in these cell types (Fig 6).
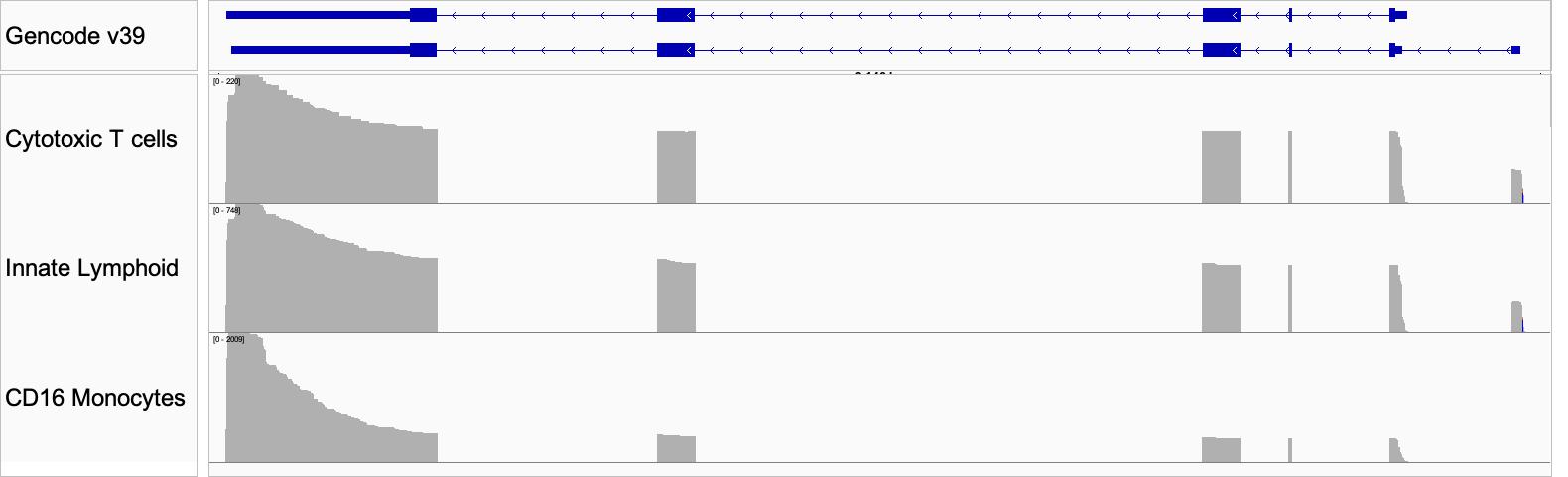
Figure 6: IGV plot of the differential isoform usage in three cell types. The top panel shows the Gencode annotation for the two isoforms identified.
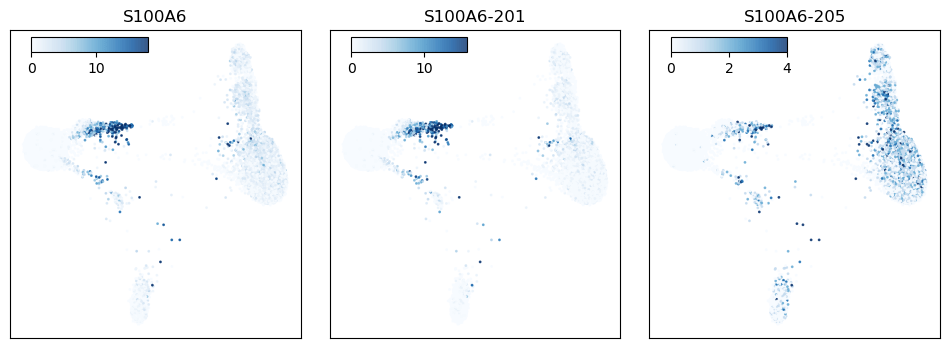
Figure 7: S100A6 expression plotted across the cell population. The left panel shows S100A6 gene expression, while the middle and right panels show S100A6 isoform expression.
Example 2: S100A6
The gene S100A6 codes for a calcium-binding protein in the S100 family that is expressed in a wide range of cell types. In these data we see the gene expressed primarily as two isoforms, with one isoform particularly highly expressed in CD16 monocytes, while other cell types show a more balanced expression profile (Fig 7).
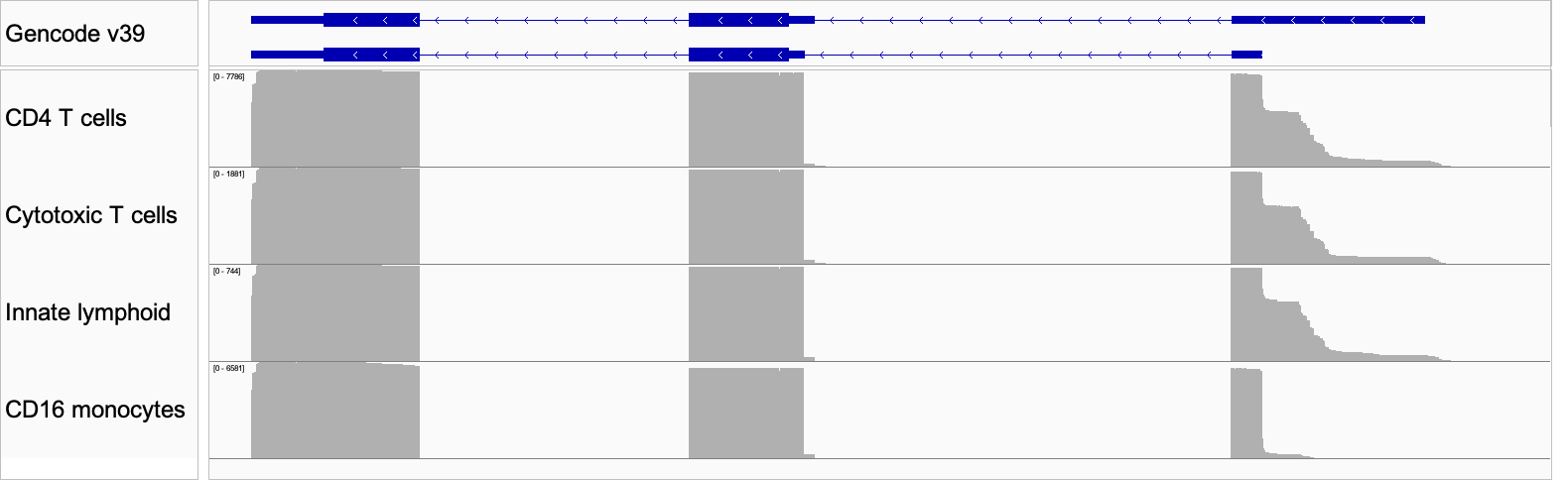
Figure 8: IGV plot of the differential isoform usage in three cell types. The top panel shows the Gencode annotation for the two isoforms identified by IsoQuant.
Again, the long-read data shows differential transcriptional start site (TSS) usage manifesting as differences in 5′ UTR sequences observed among the cell types (Fig 8). Moreover, the MAS-seq data affords us enough resolution to inspect the read pileup and see that the data are not fully explained by this combination of reference isoform annotations or indeed any other known form of this gene. This points us toward strategies for improving our isoform-calling methods, and exploring uncharted areas of the transcriptome.
Conclusion
Here we demonstrate that PIPseq is compatible with MAS-seq, expanding the validated platforms for single-cell RNA isoform sequencing. Future efforts will include cross-platform benchmarking and characterizations of single-cell platforms for RNA isoform sequencing. This early exploration highlights the transformative capability of MAS-seq and PIPseq for large-scale single-cell RNA isoform sequencing studies to efficiently identify alternative splicing in the context of development and disease.
Disclaimers
Fluent Biosciences provided PIPseq T20 3′ Single Cell RNA Kit v4.0, PIPseq starter equipment kit, and cryopreserved PBMC samples to conduct this study. PacBio provided MAS-seq kits, Revio sequencing reagents, and funding as part of a collaboration agreement.
Important note for this blog: Posts do not equal endorsements. Opinions expressed in this blog are those of the author, on behalf of the genomics group at Broad. We make every effort to ensure the accuracy of data/figures presented here but these are not peer reviewed and errors may occur from time to time.
Citations
- Clark, I. C. et al. Microfluidics-free single-cell genomics with templated emulsification. Nat. Biotechnol. 1–10 (2023) doi:10.1038/s41587-023-01685-z.
- Al’Khafaji, A. M. et al. High-throughput RNA isoform sequencing using programmed cDNA concatenation. Nat. Biotechnol. (2023) doi:10.1038/s41587-023-01815-7.
- Fontanez, K. et al. High–throughput single cell analysis with Particle–templated Instant Partitions (PIPseqᵀᴹ).
- Prjibelski, A. D. et al. Accurate isoform discovery with IsoQuant using long reads. Nat. Biotechnol. 1–4 (2023) doi:10.1038/s41587-022-01565-y.
- Inamo, J. et al. Immune Isoform Atlas: Landscape of alternative splicing in human immune cells. 2022.09.13.507708 Preprint at https://doi.org/10.1101/2022.09.13.507708 (2022).