Many individuals and families with genetic disorders are all too familiar with the concept of a “diagnostic odyssey”. It refers to the long and arduous journey to find a diagnosis, which can take years or decades, require many different tests and evaluations, and even involve dead-end incorrect diagnoses along the way. Consolidating the number of genetic tests required to reach a diagnosis could significantly reduce the length of time from symptom onset to diagnosis. Whole genome sequencing (WGS) has already been used to identify multiple variant types that previously required separate tests (eg. single nucleotide variants and copy number variants).
Identifying short tandem repeat expansions in WGS data
Expansions of short tandem repeats (STRs) are a type of variant that are responsible for many neurological disorders including, Fragile X syndrome, Friedreich ataxia, Huntington disease, myotonic dystrophy and spinocerebellar ataxia. Testing for STRs traditionally required targeted methods (triplet-repeat primed PCR or Southern blot), and while these methods are effective, they were typically ordered as a single gene analysis. Enter DRAGEN ExpansionHunter, a computational tool packaged into the Illumina DRAGEN pipeline that is capable of detecting STR expansions across many different genes using PCR-free short-read WGS data. This tool allows for a diagnostic WGS test to include reporting on STR expansions for the genes included in the tool.
The Rare Genomes Project (RGP) at the Broad Institute has been at the forefront of leveraging ExpansionHunter to identify variants responsible for rare conditions. Through their efforts, individuals with features of ataxia, myopathy, or muscular dystrophy, spanning a wide age range, have been diagnosed with STR expansions in various genes. This success story underscores the potential of computational tools like ExpansionHunter in unraveling the genetic mysteries behind rare diseases. Success of ExpansionHunter in RGP has led the Broad Clinical Laboratory to validate Illumina DRAGEN ExpansionHunter for clinical WGS testing.
Testing of DRAGEN ExpansionHunter
A cohort of 22 samples sourced from the Coriell Institute with known STR expansions in six genes (FMR1, ATXN1, FXN, HTT, C9ORF72, and DMPK) that vary by repeat size, motif, inheritance pattern, and patient sex were used. Coriell sizing was performed by Southern blot and/or PCR analysis. The normal, premutation, and full expansion repeat ranges were determined for each gene, along with a cutoff flag that would be used to flag potentially expanded alleles. These samples were run on the llumina NovaSeq 6000 system and called using DRAGEN v3.10.4 ExpansionHunter. The results of this analysis were compared to the truth data from Coriell.
As expected, sequencing read length (~150 bp) limited the ability for ExpansionHunter to call a repeat size accurately. All repeats below 150bp in length were called accurately (+/-1 repeat), whereas none of the repeats >150bp were accurately called (Figures 1a and 1b). Determining the class of expansion (normal, premutation, full mutation) is also limited for expansions beyond ~150bp. Based on these data, clinical interpretive reporting using ExpansionHunter will need to rely on the flagging cutoffs to distinguish between normal and potentially expanded alleles. However, all of the loci in this validation had flagging cutoffs that were under the 150bp limit. This may not be the case for all loci included in the ExpansionHunter caller.
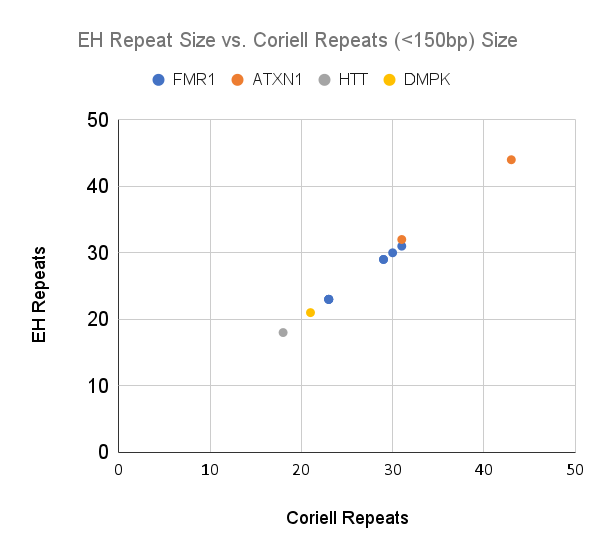
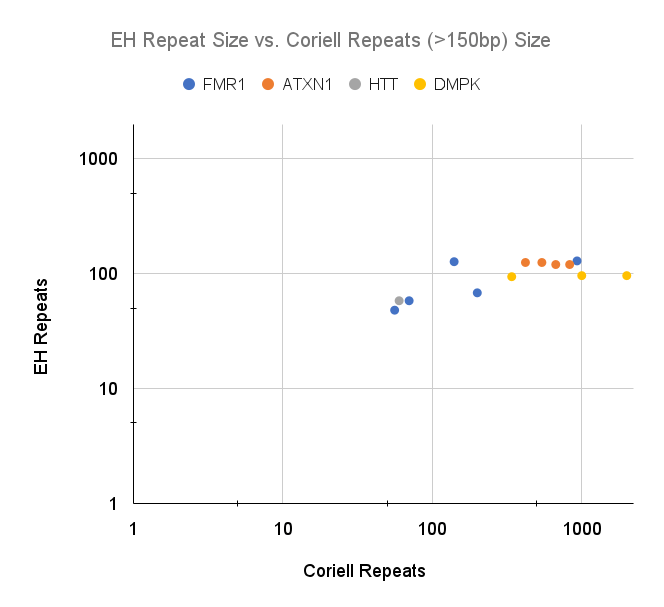
Figure 1a and 1b: ExpansionHunter repeat number compared to the number reported by Coriell. 1a) Samples with Coriell repeats <50 (<150bp) in length. 1b) Samples with Coriell repeats >50 (>150bp) in length, using a log scale for x and y. Repeats >150bp are consistently under called by ExpansionHunter.
Future directions
The integration of ExpansionHunter into clinical WGS testing is not without its challenges. Validation efforts, such as those undertaken by the Broad Clinical Laboratory, are crucial to ensuring accuracy and reliability. Validation and incorporation into clinical WGS interpretation is still in progress. These validations pave the way for incorporating ExpansionHunter calls into interpretive reports, further enhancing the diagnostic utility of WGS.
One of the key advantages of incorporating ExpansionHunter into diagnostic workflows is the expanded variant types reported. This translates to increased sensitivity, particularly for neurological disorders that may be caused by STR expansions. By reducing the need for separate test orders and streamlining the diagnostic process, patients with rare diseases can potentially receive timely and accurate diagnoses, thereby minimizing the diagnostic odyssey they often face.
In conclusion, the inclusion of ExpansionHunter in diagnostic WGS represents a significant leap forward in the field of rare disease diagnostics. Its ability to detect STR expansions with high accuracy and its integration into clinical workflows hold immense promise for improving patient outcomes and reducing the burden of the diagnostic odyssey. As advancements in computational tools continue to evolve, so too will our ability to unlock the genetic mysteries underlying rare diseases, bringing hope to patients and their families worldwide.
References:
- Dolzhenko E, et al. Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res. 2017 Nov;27(11):1895-1903. doi: 10.1101/gr.225672.117. Epub 2017 Sep 8. PMID: 28887402; PMCID: PMC5668946.
- Ibañez K, et al. Whole genome sequencing for the diagnosis of neurological repeat expansion disorders in the UK: a retrospective diagnostic accuracy and prospective clinical validation study. Lancet Neurol. 2022 Mar;21(3):234-245. doi: 10.1016/S1474-4422(21)00462-2. PMID: 35182509; PMCID: PMC8850201.
- Dolzhenko E, et al.. REViewer: haplotype-resolved visualization of read alignments in and around tandem repeats. Genome Med. 2022 Aug 11;14(1):84. doi: 10.1186/s13073-022-01085-z. PMID: 35948990; PMCID: PMC9367089.
Important note for this blog: Posts do not equal endorsements. Opinions expressed in this blog are those of the author, on behalf of the genomics group at Broad. We make every effort to ensure the accuracy of data/figures presented here but these are not peer-reviewed and errors may occur from time to time.